Bioinformatics Converter
Convert between various bioinformatics formats instantly
Paste Data
Upload File
Conversion output will appear here...
Was this tool helpful?
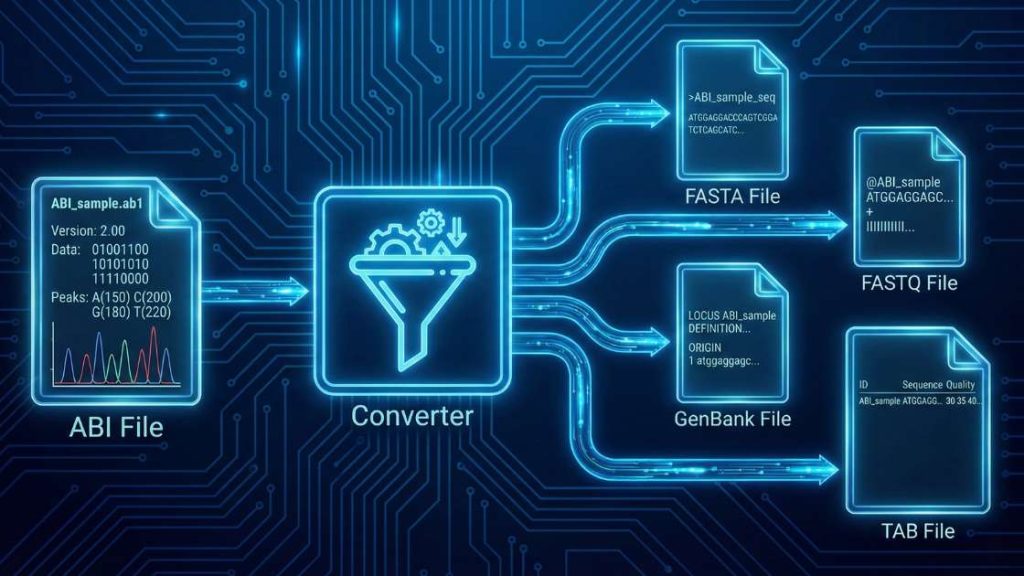
Our ABI File Format Converter is an essential web utility for molecular biologists, designed to translate raw Sanger sequencing data into universally readable formats. This tool efficiently processes binary .ab1 chromatogram files, extracting the core DNA sequence and its corresponding quality scores. By selecting the abi-trim input option, you can also automatically remove low-quality regions for cleaner, more reliable results
| Input Format | Data Type | Possible Output Formats |
|---|---|---|
abi | DNA Sequence Chromatogram (Standard Conversion) | fasta, fastq, genbank, tab |
abi-trim | DNA Sequence Chromatogram (with Quality Trimming) | fasta, fastq, genbank, tab |
How to use (step-by-step)
Follow these simple steps to convert your file in seconds.
- Upload Your File: Click “Upload File” or drag and drop your
.ab1file directly into the designated area. - Select Input and Output Formats: Use the “Input Format” dropdown to choose between
abi(standard conversion) andabi-trim(to automatically trim low-quality ends). Then, select your desired output format. - Start the Conversion: Press the “Convert File” button to begin the process. The tool will process your file instantly.
- Download Your File: Once the conversion is complete, a download link for your new file will appear. Click it to save the file to your device.
Tip: If you see an error during conversion, check the Troubleshooting Guide below—common causes and fixes are listed.
Understanding the Conversion Process
Converting your .ab1 file involves extracting and reformatting data from a complex binary structure. Here’s a breakdown of the input format, the common output formats you can choose, and the core changes that happen during the conversion.
The Input: abi & abi-trim Formats
The tool accepts one physical file type (.ab1) but interprets it in two ways based on your selection:
abi(Standard): This option treats the.ab1file as is, extracting the full, unaltered sequence and its quality scores. It is the standard binary output from Applied Biosystems (ABI) Sanger DNA sequencers and contains the raw chromatogram trace, base calls, and Phred scores.abi-trim(with Trimming): When you select this input format, you instruct the tool to perform an additional quality control step. It still reads the same.ab1file, but it will automatically trim the low-quality regions from both ends of the sequence before converting it to your chosen output format.
Because the source file is binary, you cannot view its contents in a standard text editor.
Common Output Formats & Their Uses
Your choice of output format depends on whether you need to retain the quality score information.
fastqFormat: This is the most recommended format for modern bioinformatics. It bundles the sequence identifier, the DNA sequence, and a corresponding string of quality scores for each base into a single text-based record.fastaFormat: This is the most universal format for sequence data, but it discards all quality information. Each sequence begins with a single-line description header (>), followed by the raw sequence data.tabFormat: A simple, two-column, tab-separated format where the first column is the sequence identifier and the second is the raw sequence. It also discards quality scores.
The Core Transformation Process
The converter performs these fundamental steps:
- Parses the Binary
abiFile: The tool reads the complex, multi-section binary structure of the.ab1file. - Extracts Sequence and Quality Data: It locates and extracts two key pieces of information: the final DNA base sequence and the array of per-base Phred quality scores.
- Trims Low-Quality Regions (if
abi-trimis selected): If the input format was set toabi-trim, the tool uses a modified Mott’s algorithm. It analyzes the Phred quality scores from both ends of the read and removes the regions that fall below a defined quality threshold. This process is essential for eliminating the noisy, unreliable data often found at the start and end of Sanger sequences. - Applies New Formatting Rules: It rebuilds the extracted (and possibly trimmed) data according to the text-based syntax of your chosen output format—whether that’s creating the four-line structure for
fastqor the simple header-and-sequence format forfasta.
Compatible Software
The generated output files can be used with hundreds of bioinformatics tools. The ideal software depends on your chosen format:
fastqfiles: FastQC, BWA, Bowtie2, UGENE, SnapGene, Geneious, and most next-generation sequencing (NGS) analysis pipelines.fastafiles: BLAST, Clustal Omega, T-Coffee, MAFFT, MEGA, and virtually all sequence analysis tools.
Troubleshooting Guide
Encountering an error can be frustrating, but most issues are easy to fix. Here are the most common problems you might face and how to resolve them.
General Tool Errors
- Error: “File size exceeds the limit”
- Why it happens: Your uploaded file is larger than our server’s maximum allowed size. This limit ensures fast processing for all users.
- How to fix: If your file is unusually large, please ensure it is a valid
.ab1file. For batch processing needs, please contact us for custom solutions.
- Error: “Processing timed out”
- Why it happens: The conversion is taking too long to complete, which can occur with a corrupted or unusually complex file.
- How to fix: Verify the integrity of your input file. If the issue persists with a valid file, please contact us.
- Error: “CAPTCHA validation failed”
- Why it happens: Our system uses a CAPTCHA to prevent automated bots. This error occurs if the CAPTCHA was not solved correctly or timed out.
- How to fix: Simply reload the page and solve the new CAPTCHA. If you continue to have trouble, please get in touch with our support team.
Conversion-Specific Errors
- Error: “Corrupt or unrecognized ABI file format”
- Why it happens: The uploaded file is not a valid
.ab1file, or it was corrupted during download. Our tool cannot find the specific binary markers it expects to read the chromatogram data. - How to fix: First, check the file size. If it’s 0 KB, the download was incomplete. Try re-downloading the file from your sequencing provider. As a definitive check, try opening it in a dedicated chromatogram viewer (like FinchTV or SnapGene Viewer). If it fails to open there, the file is corrupted.
- Why it happens: The uploaded file is not a valid
- Error: “Output sequence is very short after trimming”
- Why it happens: This is not a tool error, but an indication of poor-quality sequencing data. When you selected
abi-trim, the algorithm identified that most of your sequence fell below the quality threshold and was removed, leaving only a short, high-quality segment. - How to fix: Review your raw chromatogram. If you see high background noise or weak/jumbled peaks, the sequencing reaction may have failed. Consider re-sequencing the sample. To get the full (low-quality) sequence, re-process the file using the standard
abiinput format.
- Why it happens: This is not a tool error, but an indication of poor-quality sequencing data. When you selected
- Error: “No sequence data found in file”
- Why it happens: This is an issue from the sequencing machine. The
.ab1file contains the raw trace data but lacks the processed base calls (the final A, C, G, T sequence), often due to a failed analysis on the sequencer. - How to fix: This file cannot be fixed. You may need to have the sample re-sequenced or re-analyzed from the raw data. Converting this file will result in an empty output.
- Why it happens: This is an issue from the sequencing machine. The
Support Our Work
Your research matters, and we believe powerful scientific tools should be available to everyone, everywhere, without cost. This converter is built and maintained by a small team dedicated to open science.
If this tool has helped you advance your project, please consider supporting our mission. Your contribution directly funds server maintenance, ongoing updates, and the development of new free utilities for the global scientific community.
Can’t donate? You can still help! Sharing this tool with your colleagues and on social media is another powerful way to support our work. Thank you for being part of our community!
FAQ
References & Suggested Reading
This tool is built upon decades of foundational work in bioinformatics and sequencing technology. We highly recommend the following key papers and resources that have shaped the field for those interested in the underlying principles.
- Sanger, F., Nicklen, S., & Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences, 74(12), 5463–5467. https://www.google.com/search?q=https://doi.org/10.1073/pnas.74.12.5463 The seminal paper that introduced the Sanger sequencing method, which remains the gold standard for sequence validation.
- Ewing, B., & Green, P. (1998). Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Research, 8(3), 186–194. https://doi.org/10.1101/gr.8.3.186 This foundational paper describes the Phred quality score, the universally adopted metric for assessing base-call accuracy, which is crucial for our trimming algorithm.
- Cock, P. J. A., Fields, C. J., Goto, N., Heuer, M. L., & Rice, P. M. (2010). The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research, 38(6), 1767–1771. https://doi.org/10.1093/nar/gkp1137 The definitive guide to the FASTQ format, explaining how sequence and quality data are stored together—essential reading for anyone working with modern sequencing data.
- Cock, P. J. A., Antao, T., Chang, J. T., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422–1423. https://doi.org/10.1093/bioinformatics/btp163 This paper introduces the Biopython project, a vital open-source library that provides the computational engine for countless bioinformatics tools, including this one.
- Andrews, S. (2010). FastQC: A Quality Control tool for High Throughput Sequence Data. [Online]. Available: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. The official citation for FastQC, an indispensable tool for visualizing the quality of your sequencing data, and a perfect next step after using our converter.
Meet the Authors
Mahdi Morshedi Yekta
Founder & Bioinformatics Developer
Mahdi is the founder of ScienceCodons and a Medical Biotechnologist with a deep passion for computational biology. Holding an M.Sc. in Medical Biotechnology, he specializes in transforming complex biological algorithms into accessible, high-performance web tools, bridging the gap between laboratory sciences and software engineering.
Fatemeh Faryadras
Medical Biotechnologist & Researcher
Fatemeh is a Medical Biotechnologist and researcher. With extensive expertise in genetic engineering, molecular cloning, and cancer biology, she combines her rigorous laboratory background with intuitive design principles to create reliable, user-centered scientific calculators and tools.

Dr. Mosayeb Rostamian
Assistant Professor, Infectious Diseases Research Center
Certified Scientific Reviewer
Google ScholarExpert Review Process
To ensure the highest quality and scientific accuracy, this tool's underlying algorithms, formulas, and edge-case handling have been thoroughly reviewed and validated by Dr. Mosayeb Rostamian.
The "Reviewed By" badge signifies that an independent expert has evaluated the tool's logic to guarantee accuracy and alignment with the current consensus in the scientific community.