Bioinformatics Converter
Convert between various bioinformatics formats instantly
Paste Data
Upload File
Conversion output will appear here...
Was this tool helpful?
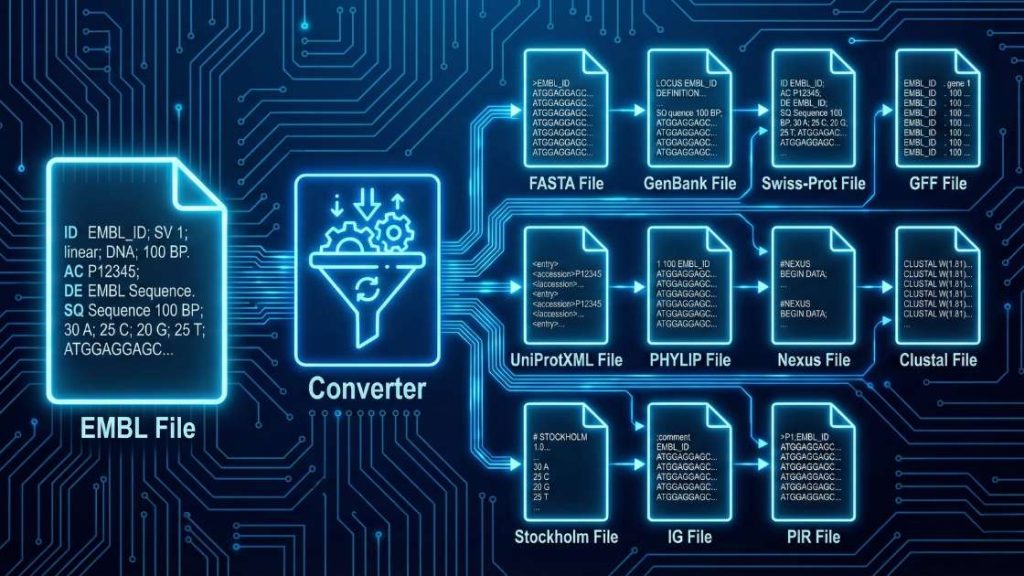
The EMBL File Format Converter is a specialized tool that converts biological sequence data from the EMBL format to various standard formats. It streamlines bioinformatics workflows by accurately parsing sequence identifiers, accession numbers, taxonomic information, and feature tables. By automating conversion, it ensures that your genomic or proteomic sequences are properly formatted for downstream applications such as multiple sequence alignment, phylogenetic analysis, and molecular docking.
| EMBL | Nucleotide/Protein Sequence | FASTA, GenBank, Swiss-Prot, GFF, UniProtXML, PHYLIP, Nexus, Clustal, Stockholm, IG, PIR |
How to use
- Upload Your File: Click “Upload File” or drag and drop your EMBL file into the designated area. You can also paste the file content using the “Paste Data” option.
- Select Your Output Format: Use the “Output Format” dropdown menu to select your desired format, such as FASTA, GenBank, or PHYLIP.
- Start the Conversion: Press the “Convert File” button to begin the process. The tool will process your data instantly.
- Download Your File: Once the conversion is complete, a download link for your new file will appear. Click it to save the file to your device.
Common Output Formats
Your choice of output format depends on the software or analysis you plan to use next.
- EMBL to FASTA Format: This is the most universal format in bioinformatics. It removes the detailed feature tables of the EMBL file, leaving only a description header (starting with >) and the raw sequence.
- EMBL to GenBank Format: A rich format similar to EMBL but used primarily by the NCBI. It retains the metadata, feature keys, and annotations.
- EMBL to PHYLIP Format: A specific format used for phylogenetic analysis software. It focuses on the alignment dimensions and sequence data rather than annotations.
- EMBL to Clustal Format: This format is for multiple sequence alignments. It displays sequences in blocks, making it easy to visualize conserved regions.
The Core Transformation Process
Regardless of the output you select, the converter performs these fundamental steps:
- Parses the EMBL Flat File: The engine scans the two-letter identifiers to distinguish between metadata (like ID and AC) and the actual sequence data in the SQ block.
- Maps Features and Annotations: If the output format supports it (e.g., GenBank), the tool maps EMBL feature keys (e.g., CDS, gene, or exon) to their corresponding equivalents in the new syntax.
- Restructures Sequence Strings: It removes the numbers and spaces typically found in the EMBL SQ block and reformats the sequence into the required width or continuous string required by the target format.
Troubleshooting Guide
Error: “AttributeError: ‘NoneType’ object has no attribute id. Why it happens: This usually indicates that the EMBL header is missing the mandatory “ID” line or that the ID line is malformed, preventing the engine from identifying the sequence record. How to fix: Check the first line of your file. Ensure it begins with “ID”, followed by the appropriate identification string and the sequence length.
Error: “ValueError: Sequences must be the same length for this format.” Why it happens: This occurs when you attempt to convert an EMBL file containing multiple sequences into a strict alignment format (like PHYLIP or Clustal) where the sequences are not of equal length. How to fix: If you are creating an alignment, ensure sequences are pre-aligned with gaps. If not, choose a non-alignment format like FASTA or GenBank.
Error: “StopIteration” during parsing. Why it happens: This error suggests the file is truncated or the “//” terminator at the end of the EMBL record is missing, causing the parser to reach the end of the file unexpectedly. How to fix: Ensure your file is complete and that every individual record ends with the standard “//” line on its own row.
If your problem is not listed here, let us know. Please help us improve the tool by reporting the issue.
Support Our Work
We are dedicated to providing free, high-quality computational tools for the scientific community. Maintaining these resources requires time and server costs. If this EMBL converter has helped your research, please consider supporting our work. Your contributions help us keep these tools up to date, add new formats, and ensure they remain accessible to researchers worldwide.
References
Peter J. A. Cock, Tiago Antao, Jeffrey T. Chang, Brad A. Chapman, Cymon J. Cox, Andrew Dalke, Iddo Friedberg, Thomas Hamelryck, Frank Kauff, Bartek Wilczynski, Michiel J. L. de Hoon, Biopython: freely available Python tools for computational molecular biology and bioinformatics, Bioinformatics, Volume 25, Issue 11, June 2009, Pages 1422–1423, https://doi.org/10.1093/bioinformatics/btp163
Meet the Authors
Mahdi Morshedi Yekta
Founder & Bioinformatics Developer
Mahdi is the founder of ScienceCodons and a Medical Biotechnologist with a deep passion for computational biology. Holding an M.Sc. in Medical Biotechnology, he specializes in transforming complex biological algorithms into accessible, high-performance web tools, bridging the gap between laboratory sciences and software engineering.
Fatemeh Faryadras
Medical Biotechnologist & Researcher
Fatemeh is a Medical Biotechnologist and researcher. With extensive expertise in genetic engineering, molecular cloning, and cancer biology, she combines her rigorous laboratory background with intuitive design principles to create reliable, user-centered scientific calculators and tools.

Dr. Mosayeb Rostamian
Assistant Professor, Infectious Diseases Research Center
Certified Scientific Reviewer
Google ScholarExpert Review Process
To ensure the highest quality and scientific accuracy, this tool's underlying algorithms, formulas, and edge-case handling have been thoroughly reviewed and validated by Dr. Mosayeb Rostamian.
The "Reviewed By" badge signifies that an independent expert has evaluated the tool's logic to guarantee accuracy and alignment with the current consensus in the scientific community.